
Research Models
|
Toni Ahtoniemi
The Ultimate Ice Bucket Challenge
Finding more effective ways of studying ALS in animal models
The ice bucket challenge heard round the world (even way up here in Finland) has helped raise awareness and over US$110 million (by last count) for amyotrophic lateral sclerosis (ALS) research. The bigger challenge will be finding more effective treatments, a cure or a way to prevent this complex neurodegenerative disorder.
ALS or Lou Gehrig’s disease is one of the worst diseases you could possibly imagine. The disease burns out motor neurons, causing the brain to lose its ability to control muscle movement. People afflicted with this chronic, ultimately fatal condition lose their ability to walk, speak, breathe and feed themselves. In the end, they live in a locked-in state, watching their bodies waste away.
Treatment options are slim. The only approved drug, Riluzole, prolongs disease progression a mere four months on average, and disease wins out in the end. Most people with ALS die within two to five years following diagnosis. As with other neurodegenerative diseases like Alzheimer’s or Parkinson’s, the mechanisms of ALS are multifactorial, and have been challenging to translate into effective therapies.
Only about 10% of ALS cases are genetic (the rest occur sporadically), yet it has been in this small subset of individuals that researchers (including myself) have spent most of their time trying to understand what triggers the disease and how to overcome it. The most well-studied gene is the human SOD1 gene, which expresses a protein that helps break down toxic oxygenated molecules called superoxide radicals. At least 170 mutations in the SOD1 gene have been linked to inherited cases of ALS. There are also 20 or so other genes also linked to ALS that have been identified in recent years.
Popular Transgenic Model
A transgenic mouse model carrying a human mutant form of enzyme superoxide dismutase (SOD1) that causes similar ALS symptoms as those found in humans, has been used extensively to study ALS and test new drug candidates. We have tested dozens of compounds in the SOD1 model, for instance, to see if they have any effect in delaying symptoms or improving motor performance. Typically, we follow the disease progression with several motor behavioral tests including rotarod, grip strength and open field as well as with basic monitoring such as clinical score and body weight. In addition to the SOD1 models, there are also other rodent models available including naturally occurring genetic models and transgenic mice with mutations with FUS or TDP-43.
The drawback with all these models, including the SOD1 model, is that they represent very narrow subsets of the entire field of human ALS cases, making them less than perfect canaries in the proverbial coal mine. For practical reasons, they also manipulate certain aspects of ALS so that scientists can study them. For instance, one catch in the SOD1 model is the high overexpression of the mutant gene associated with ALS, fast-forwarding the mice into disease.
What all this means is that developing drug candidates that target an enzyme or pathway associated with one of these genetic anomalies may work in the animal model that has that mutation, but not so well in human clinical testing due to the diversity of the genes (and suspected environmental factors) that seem to be driving ALS.
New Directions in Research
With this in mind, there has been a lot of discussion about how these models should be used and which models are the most appropriate. To bring more value to the studies, drug developers have been testing their compounds in multiple models; a drug that has performs well in multiple studies has a better chance of showing efficacy in clinical studies.
Researchers are also making strides in catching ALS sooner—the time when treatments are more likely to work—and in translating ALS from mice to men by using motion-capture computer-generated imagery. Fine motor kinematic analysis—the geometry of mice in motion—and specific algorithms enable scientists to detect subtle phenotypic chances with earlier and more sensitive detection compared to traditional movement analysis. Our lab began using this kind of kinematic analysis about a year now to model and measure subtle movement and coordination in rodents with Huntington’s Disease, see Eureka post, and we began applying it to the SOD1 model about six months ago (see image below).
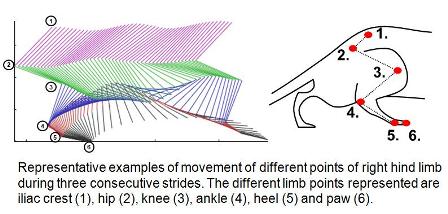
Being able to catch subtle changes in motor control during the pre-symptomatic phase has enabled us to identify a lot of different endpoints (such as tail height, gait and limb coordination among the 100 parameters available) that we hope will guide drug development in ways that weren’t possible before.
Traditionally, the ALS models have focused disproportionately on survival and on the later stages of disease, when the twitching, tremors and loss of motor control become visible and the damage so extensive that it’s nearly impossible to attack it therapeutically. With kinematic analysis we can actually look at the pre-symptomatic change and focus on subtle changes that occur sooner to see if drug compounds have an impact.
With time, we might have better success in the clinic, and perhaps a cure for ALS, which affects 350,000 people worldwide. But first we’ll need to further refine our preclinical models. Translating such a complex disease is the ultimate ice bucket challenge.