.jpg?crop=true&keep=c&q=80&color=ffffffff&u=8anjqk&w=1400&h=500)
Discovery
|
Taneli Heikkinen
Hope for Huntington’s Disease
An experimental gene therapy is able to slow the progression of symptoms in patients, a long-awaited glimmer of hope for people afflicted with this deadly and debilitating disease
Huntington’s disease (HD) is a genetic neurodegenerative disorder caused by a mutation in the huntingtin gene on chromosome 4. In this mutation, a specific CAG trinucleotide repeat has an abnormally large number of repeats, resulting in the production of a toxic form of the huntingtin protein. When the number of these CAG repeats exceeds 39 in an individual, the person will develop HD. The average age of onset is approximately 40. HD is typically inherited from an affected parent who carries the mutation. The disease symptoms include progressive motor dysfunction, cognitive decline, and psychiatric symptoms. Most patients die within 15 years after symptoms surface. The prevalence of HD is 3 / 100,000 in Finland and 6 / 100,000 in Europe. However, there are locations, for example, in South America, where the incidence is even higher—55 / 100,000.
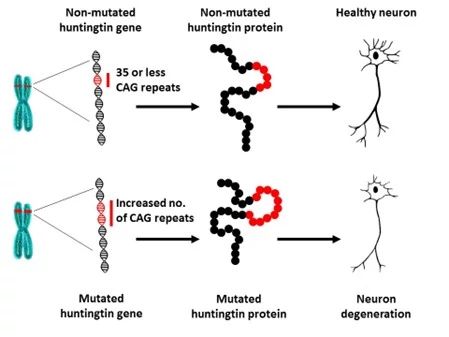
Until now, no treatments have been effective in showing the progression of symptoms, let alone curing the disease in HD patients. Fortunately, the tide is turning. On September 24, the gene therapy company uniQure announced findings from a clinical trial that showed HD patients had a 75% slower disease progression after receiving their therapy. The results were demonstrated in trial participants who received the higher dose of treatment.
uniQure’s gene therapy, named AMT-130, uses a specially designed harmless virus called AAV5 to deliver microRNA directly into the brain. In the case of HD, the area of the brain targeted by the drug is the striatum, which is associated with the execution of both simple and more complex tasks. AMT-130 targets the huntingtin mRNA to reduce production of both mutant and normal huntingtin protein. Prior to testing in people, studies found that targeting human HTT mRNA is effective in lowering the soluble mutant HTT protein and reducing the amount of HTT aggregates in the brain in a relevant murine model of HD.
Altogether, this is a remarkable finding from uniQure and may prove to be revolutionary on the long road toward a cure for HD. To offer patients a longer and healthier life would be nothing short of transformative. However, the trial is still ongoing, the treatment remains experimental, and certain open questions remain unanswered.
Still, this holds more promise than any treatment for HD so far. The findings show that HD progression can be slowed down in patients. This outcome holds promise for other treatment candidates as well, especially those focused on lowering levels of huntingtin. The whole HD community is rightfully excited about this, and for good reasons.
Success stories don’t come often, not on a monthly or yearly basis. Given the complexity of genetic diseases, the slow pace of even research is understandable. But as we can see here, successes do occur. This is a good example of the importance of perseverance, great science, and the realization that research matters.
Taneli Heikkinen, PhD, is a Senior Scientist at Charles River Laboratories’ site in Kuopio, Finland, which specializes in neuroscience. HD is among the many neurological conditions that Kuopio studies.



