Biologics
|
Matthew Pennington, PhD
SARS-CoV-2 Mutations Complicate COVID-19 Pandemic Response
The variants do not appear to impact vaccine efficacy, but they will likely drive up cases
The genome of a virus, composed of either RNA or DNA, is the set of directions that a virus needs to reproduce itself. It encodes amino acids, forming viral proteins that lead to the production of new copies of the virus. The genome itself must also be copied, usually by viral polymerases, and passed to the new copies. However, these polymerases can be error prone, spontaneously mutating to cause the wrong nucleic acid to be incorporated into the genome.
These mutations can be identified by sequencing samples collected from patients, essentially reading the letters of the virus’s genome, and comparing how different the samples are from each other. There are several different types of mutations that might occur, which all have different effects on the sequence of the resulting protein (Figure 1). Identifying the formation of these mutations can allow us to trace back when and where new virus variants have emerged and how they are spreading around the world. It can also give us insight whether the biology of the virus is changing.
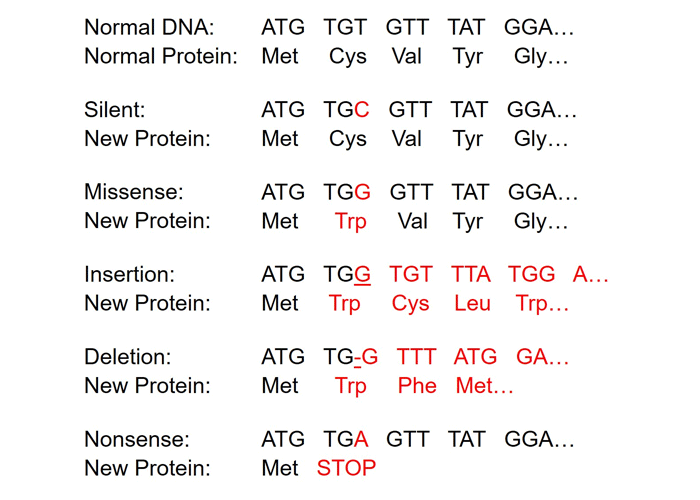
Figure 1: Types of mutations, shown for a DNA virus. Amino acids are encoded by sets of three base pairs called codons. This normal DNA sequence encodes the amino acids Met-Cys-Val-Tyr-Gly, and on, resulting in a protein. The type of mutation affects the resultant protein: (1) Silent mutations change the DNA sequence, but do not result in an amino acid change. (2) Missense or substitution mutations result in a change of one amino acid, without affecting the rest of the sequence. These may change the structure of the protein at the site at which they occur. (3) Insertion of an extra nucleotide or (4) deletion of a nucleotide may result in a frameshift mutation, resulting in an entirely different, often non-functional protein. Insertions or deletions may also add or remove entire amino acids, without resulting in a frameshift. (5) Nonsense mutations can occur as a result of any of these mutations and result in the introduction of a Stop signal, ending production of the protein early. Viruses containing RNA genomes are susceptible to the same types of mutations (not depicted here).
Mutations may be good, bad, or indifferent to the virus. Natural selection and evolutionary pressures will drive their maintenance or loss in a population. If the mutation does not change the amino acid code or the structure of the protein in a meaningful way, likely there will be no effect on the virus. The mutation may harm the virus if it results in a nonfunctional protein, causing these variants to be outcompeted and lost from the population. The mutations may also help the virus, conferring some new or enhanced ability compared to the original form. Natural selection will drive expansion of these variants as they outcompete the other strains (Figure 2).
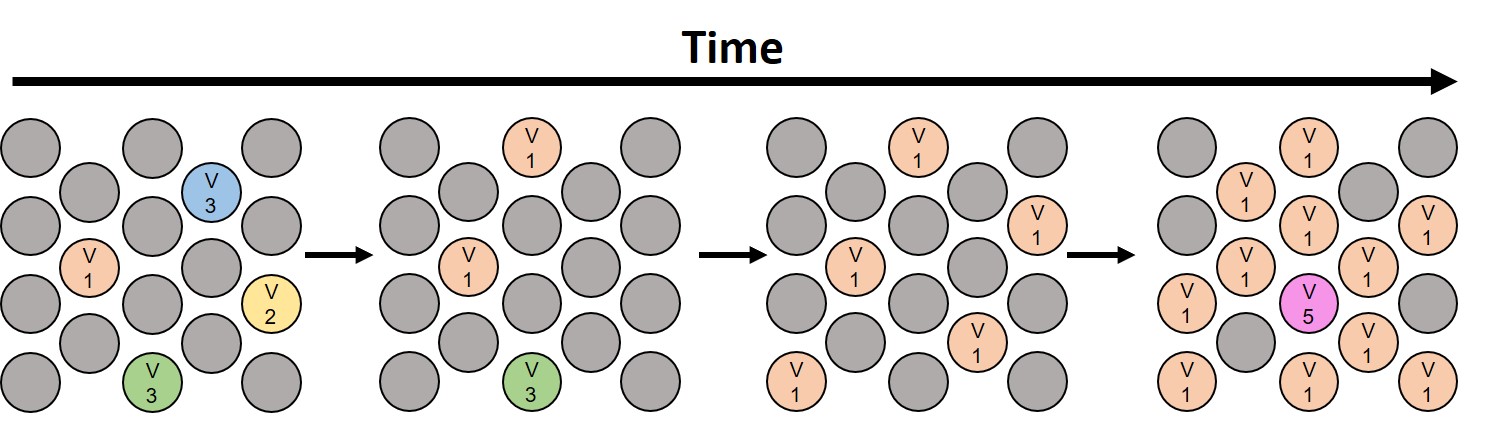
Figure 2: Emergence of a viral variant in a population. A population is initially dominated by one form of a virus (here depicted in gray). Random viral variants exist at low levels and/or spontaneous develop. Over time, one variant gains a competitive advantage over the initial form of the virus. Natural selection selects for this variant, allowing it to spread and outcompete the initial virus. Eventually, this variant may coexist with the initial form, or even supplant it as the dominant virus. New variants can continue to emerge and may themselves rise to dominance.
There are many potential consequences if a new variant of a virus takes hold. It may gain the ability to spread more quickly, may cause more severe disease, and may be more deadly. If the changes are large enough, it may allow the virus to evade diagnostic tests to identify it. Variants may be more resistant, or even entirely immune to therapies. The variant may also gain the ability to escape recognition by the immune system, meaning vaccines and natural immunity may not be effective against them.
Naming the COVID-19 Mutants
SARS-CoV-2, the virus which causes COVID-19, can, like all viruses, mutate. Since December 2020, at least three new variants have been identified and are widely circulating. Naming these variants is complicated. No formalized naming system yet exists for COVID variants, and so the identified variants tend to each have multiple names. In the news, they have been referred to by the country in which they were first identified, for example, the UK variant. However, this method is not terribly useful. First, as with the naming of virus species, it is typically preferred to avoid using country names for geopolitical reasons. Second, variants are not necessarily first identified in the country in which they first emerged. Third, multiple variants may exist in or have originated from the same country at the same time, raising the question of, for example, which UK variant? The World Health Organization is holding meetings to develop a naming convention. Here, we will use the most widely recognized name for each variant.
So what has changed?
The B.1.1.7 strain (also called 20I/501Y.V1 and VOC 2021/01) was the first major variant to be identified. It originally emerged in the United Kingdom and was identified in samples collected in late September 2020. By January 2021, it was responsible for more than half of the new infections in the UK. It has also spread to more than 60 countries, including in at least 20 states of the USA. And preliminary studies suggest the B.1.1.7 variant might be more deadly than the original. The data out of the UK is based on a handful of cases and scientists cautioned that the findings are far from conclusive.
B.1.1.7 contains 17 distinct mutations compared to the original form of SARS-CoV-2. These have been extensively illustrated by the New York Times. Eight of these mutations occur in the viral spike protein, which is used to gain entry into host cells. These are of most interest and most likely to change the biology of the virus.
Perhaps the most important mutation is the substitution of a tyrosine (Y) in place of asparagine (N) at position 501 of the spike protein (referred to as the N501Y mutation). This mutation occurs in the receptor binding domain of the spike protein. This region is key to properly latching onto the ACE2 receptor on host human cells in order to gain entry. This mutation appears to make the virus grab onto the receptor much more tightly, increasing the chance of infection.
B.1.1.7 has several other interesting mutations. Preliminary studies suggest that the deletion of amino acids 69-70 and 144-145 from the spike protein may change the way antibodies recognize the virus. The substitution of a histidine (H) instead of proline (P) at position 681 (P681H) occurs in the cleavage site of the spike protein, the site at which it must be cut apart to allow the virus to enter cells. This mutation appears to make this process more efficient, potentially making infection easier. Finally, there is a nonsense mutation that introduces an early stop codon into the open reading frame 8 (Orf8) of the virus, resulting in a short and presumably non-functional protein. The importance of this mutation is not understood.
The B.1.351 strain (also called 20H/501Y.V2) was originally detected in October 2020 in South Africa. It has spread to ~20 other countries, including Zambia where it is the dominant strain. It also contains eight mutations in the spike protein. It shares the N501Y mutation with B.1.1.7. However, the other mutations are different, and it does not contain the deletions, suggesting it emerged independently from B.1.1.7. The most interesting mutation in this strain is the substitution of a lysine (K) for glutamic acid (E) at position 484 (E484K), which may also affect the ability of antibodies to recognize this virus.
The P.1 strain was found in Manaus, Brazil and also identified by Japanese health officials in four travelers returning from Brazil. This shares the N501Y and E484K mutations with the B.1.351 strain, but also has 17 unique mutations, including in the receptor binding domain, supporting the hypothesis that it emerged independently of both of the other strains. More variants are being identified as more samples are sequenced. For example, a new variant was identified in California with an L425R mutation in the receptor binding domain of the spike protein, though the implications of this mutation are not yet understood.
What are the impacts of these mutations?
These variants raise two key questions: (1) Do these mutations change how the virus spreads and its severity, and (2) will the vaccines still work? Mutations in the receptor binding domain, and primarily the N501Y mutation, appear to be associated with an increase in transmissibility. One preliminary study suggested that B.1.1.7 is 56% more transmissible than pre-existing forms of SARS-CoV-2. The good news is that none of these strains appear to be associated with more severe disease or more deaths. However, it is likely that more people will die as these strains spread, not due to an inherently more deadly variant, but rather simply due to an increase in case numbers and additional strain on healthcare systems.
The COVID vaccines currently in use primarily work by stimulating the immune system to produce a diverse array of antibodies, known as a polyclonal response, to bind to and neutralize the virus. If the mutations change the virus in such a way that those antibodies cannot bind to it, then it is possible the vaccines will not be effective. It appears that the N501Y mutation does not allow the virus to evade detection. As mentioned above, preliminary studies suggest that the 69-70 deletion, the E484K mutation, and other mutations may allow the virus to avoid recognition in cell culture tests. However, the polyclonal nature of the response means that that while these variants may escape detection by some antibodies, they should still be recognized by others. Complete vaccine escape is unlikely. This means that the vaccines may be less effective against certain variants but should still provide a level of protection. An active and aggressive area of research is to determine how much protection the vaccines provide against these variants and our understanding may change as new data comes in. This week the maker of one of the RNA vaccines currently being distributed said it provided protection against both B.1.1.7 and B.1.351 strains but the company is developing a new vaccine against the B.1.351 as a precaution.
Though these variants seem like they appeared suddenly, their emergence was expected as viruses constantly change over time. It is only in the past few months that our sequencing capabilities have begun to scale up. As more samples are sequenced, more variants will be identified. The challenge for scientists and public health officials is to understand the mutations present in these variants compared to the original SARS-CoV-2 and how they impact the behavior of the virus. In the meantime, the advice for handling these variants is the same as it has always been — wear a mask, socially distance, wash your hands, and get vaccinated when it is available.
In the original version of this article, posted Jan 25, 2021, we noted that some of the specific amino acid changes observed with the SARS-CoV-2 variants potentially impacted their ability to be detected by antibodies in cell culture testes. However, at the time it did not appear that these mutations would impact vaccine efficacy. Science moves quickly and new data has come in. Recently, a trial of a major vaccine candidate was halted in South Africa as it was found that the vaccine did not provide protection against mild or moderate illness caused by the B.1.351 variant. It does appear to still protect against severe disease, suggesting, and as we noted, that variants may prove to be partially resistant to our vaccines, but were unlikely to be fully resistant. So far, the data suggest that the currently approved mRNA vaccines remain effective, though this may also change as our understanding of these mutants change.
We also noted that the B.1.1.7 variant, first identified in the United Kingdom, while it is more transmissible, did not appear to cause more severe disease or inherently be more deadly. A preliminary study has suggested that perhaps this variant is associated with an increased risk of death. Additional studies are needed to understand these existing variants. Nevertheless, our suggestions to get vaccinated when it is your turn, wear a mask, wash your hands, and social distance remain the best advice to slowing the spread of these variants.